Identification of growth trait related genes in a Yorkshire purebred pig population by genome-wide association studies
Article information
Abstract
Objective
The aim of this study is to identify genomic regions or genes controlling growth traits in pigs.
Methods
Using a panel of 54,148 single nucleotide polymorphisms (SNPs), we performed a genome-wide Association (GWA) study in 562 pure Yorshire pigs with four growth traits: average daily gain from 30 kg to 100 kg or 115 kg, and days to 100 kg or 115 kg. Fixed and random model Circulating Probability Unification method was used to identify the associations between 54,148 SNPs and these four traits. SNP annotations were performed through the Sus scrofa data set from Ensembl. Bioinformatics analysis, including gene ontology analysis, pathway analysis and network analysis, was used to identify the candidate genes.
Results
We detected 6 significant and 12 suggestive SNPs, and identified 9 candidate genes in close proximity to them (suppressor of glucose by autophagy [SOGA1], R-Spondin 2 [RSPO2], mitogen activated protein kinase kinase 6 [MAP2K6], phospholipase C beta 1 [PLCB1], rho GTPASE activating protein 24 [ARHGAP24], cytoplasmic polyadenylation element binding protein 4 [CPEB4], GLI family zinc finger 2 [GLI2], neuronal tyrosine-phosphorylated phosphoinositide-3-kinase adaptor 2 [NYAP2], and zinc finger protein multitype 2 [ZFPM2]). Gene ontology analysis and literature mining indicated that the candidate genes are involved in bone, muscle, fat, and lung development. Pathway analysis revealed that PLCB1 and MAP2K6 participate in the gonadotropin signaling pathway and suggests that these two genes contribute to growth at the onset of puberty.
Conclusion
Our results provide new clues for understanding the genetic mechanisms underlying growth traits, and may help improve these traits in future breeding programs.
INTRODUCTION
Growth rate is an important factor in pig breeding programs as it is directly associated with economic benefits [1]. Growth rate is typically represented by traits such as average daily gain (ADG) and days to 100 or 115 kg (D100 or D115). In order to design better breeding programs and to improve production efficiency, it is important to understand the genetic determinants controlling these traits [2]. According to data in the pig quantitative trait locus (QTL) database, 15108 QTLs from 537 publications have been reported to be associated with 600 different traits [3]. Of these, 1315 QTLs are associated with growth traits, including 563 QTLs for ADG and 18 QTLs for days to different body weights (http://www.animalgenome.org/cgi-bin/QTLdb/SS/index, Apr 29, 2016). QTL data suggests that growth traits are controlled by many loci [1], but the causative genes or single nucleotide polymorphisms (SNPs) are difficult to identify due to the low resolution and complex architecture of most QTLs [4,5].
Because of the availability of high-throughput genotyping platforms for SNPs, genome-wide association studies (GWAS) has become a powerful method for dissecting the genetic basis underlying traits in both humans and animals. Since the development of the Illumina PorcineSNP60 Genotyping BeadChip, GWAS has been performed in commercial pig populations, revealing strong associations for economically important traits including body composition [6], meat quality [7], and reproductive performance [8]. Many GWAS have been performed in different pig populations in order to identify genes controlling production traits [1,2,9]. In a previous study, we performed a GWAS to analyze production traits in a Duroc male pig population and identified 14 candidate genes [2]. Two models were used to identify SNPs associated with feed conversion ratio, by which 13 significant SNPs were identified [9]. Another GWAS identified 127 significant SNPs and 102 suggestive SNPs associated with ADG in Italian Large White pigs [1]. When GWAS was used to examine residual feed intake and ADG in two extremely divergent purebred Yorkshire lines, significant SNPs were identified on several chromosomes including Sus scrofa chromosome (SSC)3, SSC5, SSC6, SSC7, SSC13, SSC14, and SSC15 [10].
To identify new candidate genes involved in pig growth, we performed a GWAS in a purebred Yorkshire pig population containing approximately 600 individuals for the growth traits D100, ADG100, ADG115, and D115, using the GeneSeek-Neogen Porcine SNP80 BeadChip.
MATERIALS AND METHODS
Source population and phenotypes
Five hundred and sixtytwo commercial American Yorkshire female pigs were raised and performance-tested at the Beijing Breeding Swine Center (Beijing, China). All animals were born at the end of 2013 and raised under identical standard conditions. No open wounds or other signs of illness, injury, or abnormal behavior, were observed at any time. Sample (ear tissue) collection was conducted based on procedures described previously [2]. The protocol for ear tissue collection was approved by the Animal Welfare Committee of the China Agricultural University (Approval number: XK257).
Four traits were recorded for individual pigs: D100 and D115 (days to 100 or 115 kg of body weight), and ADG100 and ADG115 (ADG between 30 and 100 or 115 kg). Phenotypes were collected using the Osborne FIRE Pig Performance Testing System (Osborne, KS, USA) at the Beijing Breeding Swine Center (Beijing, China). D100, D115, ADG100, and ADG115 were used in the subsequent genome-wide association analysis.
Genotyping and quality control
DNA was extracted from ear tissue using phenol-chloroform [11]. DNA quality and quantity were assessed using a NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA). Genotyping was conducted using the GeneSeek-Neogen Porcine SNP80 BeadChip (Beijing Kangpusen Biological Technology Co., LTD, Beijing, China). Data mining was performed in our laboratory.
To reduce false-positive associations resulting from genotyping, we controlled our SNP analysis with a genotyping call rate ≥90% (individuals and SNP) and a Hardy–Weinberg equilibrium p≥10−6. Considering that rare SNPs have lower statistical power, SNPs with a minor allele frequency (MAF) ≥3% were selected for further analysis. All SNPs located on the chromosome Y were removed.
Genome-wide association studies analysis
The GWAS were performed as described previously, using our Fixed and random model Circulating Probability Unification method (FarmCPU) [12]. Bonferroni multiple testing was used to correct the genome-wide significant (0.05/N) and suggestive (1/N) thresholds, where N is number of SNPs used in the analysis. The quantile–quantile (Q–Q) plot, which is a commonly used tool for scanning the population stratification in GWA studies, was always implemented in the test [13]. Principle component analysis was performed by Plink v1.07 [14].
Functional analysis
To identify functionally plausible candidate genes, we searched for the closest annotated gene within a 1 Mb region centered on each significant or suggestive SNP in the pig reference genome assembly. The gene identifiers were used to identify the orthologous mouse genes in Ensemble BioMart (http://www.ensembl.org/biomart/martview). Using the mouse orthologs, gene ontology (GO) analysis was carried out using the DAVID Bioinformatics Resources version 6.7 (http://david.abcc.ncifcrf.gov/) [15]. A gene network analysis, using the names of the candidate genes, was performed in GeneMANIA (http://genemania.org/).
RESULTS
Phenotype and single nucleotide polymorphisms data summary
Phenotype data for the four traits of interest are summarized in Table 1. All traits were approximately normally distributed. After a series of filtering steps, 2,878 SNPs with no physical location, 14 SNPs located on chromosome Y, 342 SNPs with call rate below 90% and 7,146 SNPs with MAF lower than 3% were removed. In total, 54,148 SNPs were available for GWA analysis (Table 2). The average physical distance between two neighboring SNPs on the same chromosome was approximately 45.9 Kb, ranging from 31.2 Kb (SSC12) to 61.5 Kb (SSCX). Based on the length of each chromosome in the USDA-MARC v2 (A) linkage map, the average genetic distance between adjacent SNPs was 0.043 cM, with a maximum of 0.056 cM (SSC17) and a minimum of 0.028 cM (SSC1) (Table 2).
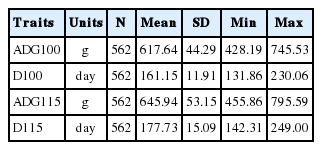
Descriptive statistics for growth traits in a Yorkshire pig population
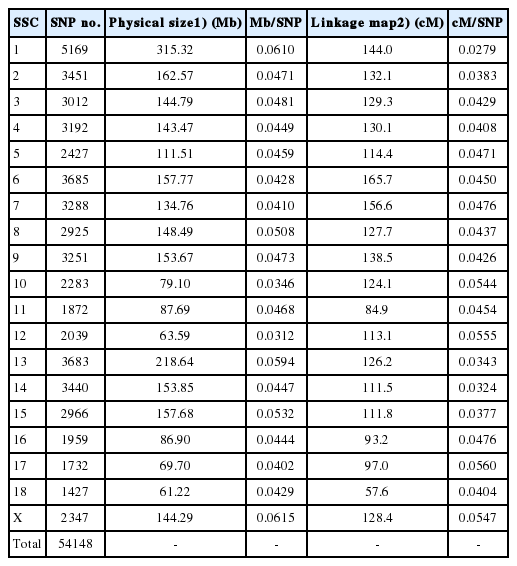
Distribution of SNPs after quality control and average distance between adjacent SNPs on each chromosome
Significant single nucleotide polymorphisms
Based on multidimensional scaling analysis, no obvious population structure was observed in our population. The p-value profiles (−log p-value) of all SNPs associated with the four traits are shown in Figure 1 (analyzed using FarmCPU). The genome-wide significant or suggestive SNPs associated with the four growth traits are shown in Table 3. In total, 6 genome-wide significant and 12 genome-wide suggestive SNPs were associated with the ADG100, ADG115, D100, and D115 traits.
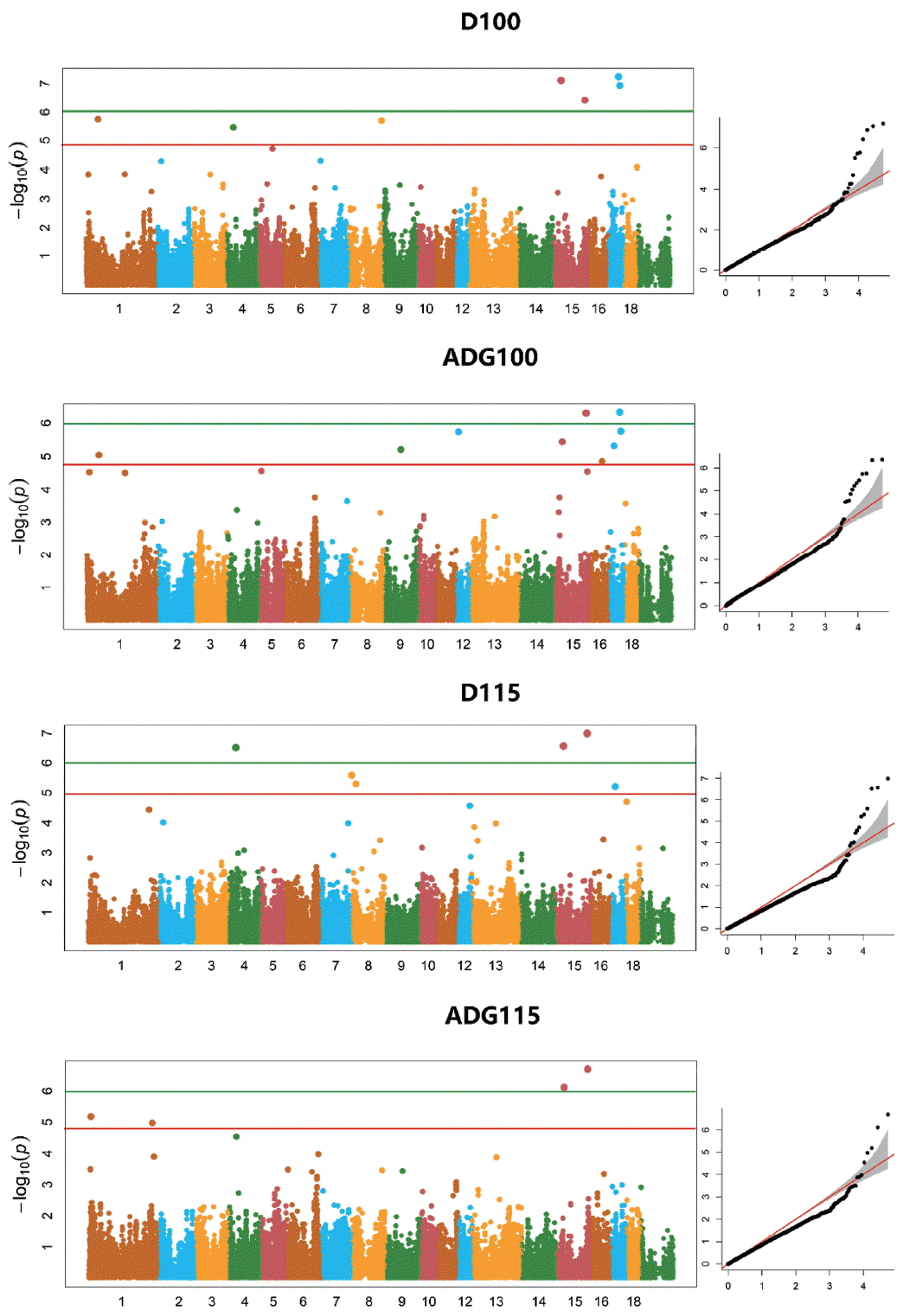
Manhattan plots for the four growth traits examined in the genome-wide association study using Yorkshire pigs. The quantile-quantile plots on the right show the observed versus expected log p-values.
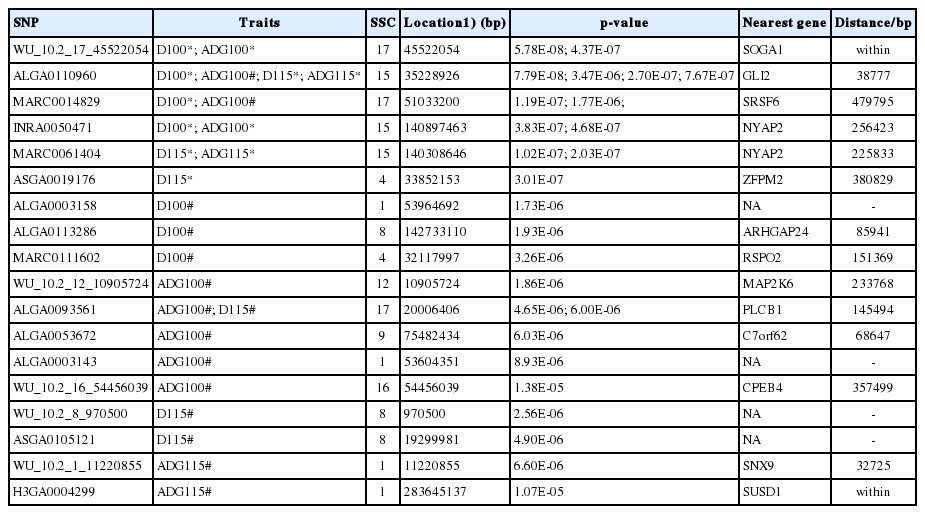
Significant and suggestive SNPs associated with growth traits
Five of the six significant SNPs associated with multiple traits. SNP ALGA0110960 located on SSC15 associated with four traits. SNPs WU_10.2_17_45522054 and INRA0050471 both associated with the D100 and ADG100 traits. MARC0014829, located on SSC17, associated with D100 and ADG100, whereas MARC0061404, located on SSC15, associated with D115 and ADG115. The suggestive SNP ALGA0093561, located on SSC17, associated with ADG100 and D115. Finally, three suggestive SNPs associated with D100; a significant and two suggestive SNPs associated with D115; four suggestive SNPs and two suggestive SNPs associated with ADG100 and ADG115, respectively (Table 3).
Growth-related candidate genes
To identify growth-related candidate genes, the annotated genes closest to significant and suggestive SNPs were identified and then evaluated through literature mining to assess their biological functions. The search yielded nine candidate genes potentially associated with D100, ADG100, D115, and ADG115. These genes are: suppressor of glucose by autophagy (SOGA1), GLI family zinc finger 2 (GLI2), neuronal tyrosine-phosphorylated phosphoinositide-3-kinase adaptor 2 (NYAP2), zinc finger protein multitype 2 (ZFPM2), rho GTPASE activating protein 24 (ARHGAP24), R-Spondin 2 (RSPO2), mitogen activated protein kinase kinase (MAP2K6), phospholipase C beta 1 (PLCB1), and cytoplasmic polyadenylation element binding protein 4 (CPEB4). The result from a network analysis of the candidate genes is shown in Figure 4. Most of the related genes in the network also exhibit functions associated with growth traits, either directly or indirectly. For example, glycerol-3-phosphate acyltransferase 3 (AGPAT9) plays an important role in adipocyte differentiation and triacylglycerol biosynthesis [16].

Gene network showing the 9 candidate genes and their potential interactions.
DISCUSSION
GWAS is a powerful method to identify the mutations or genes underlying complex traits in domestic animals [4]. In the pig, GWAS has been performed to identify candidate genes for traits related to production [2], body composition [6], reproduction [17], and immunity [18]. In this study, GWAS was performed for four growth traits (ADG100, ADG115, D100, and D115), for which 6 genome-wide significant and 12 genome-wide suggestive SNPs were identified. It is well known that the accuracy of GWAS can be compromised by population stratification [19]. In contrast to humans, the relatively simple genetic background found in domestic animals makes it easier to identify loci that are truly responsible for corresponding traits [4]. The Yorkshire pigs used in this study had a similar genetic structure because they originated from a single farm. In addition, multidimensional scaling and Q-Q plots showed no obvious population structure in our population (Figures 1 and 2). This suggests that GWAS yielded valid results in our study.
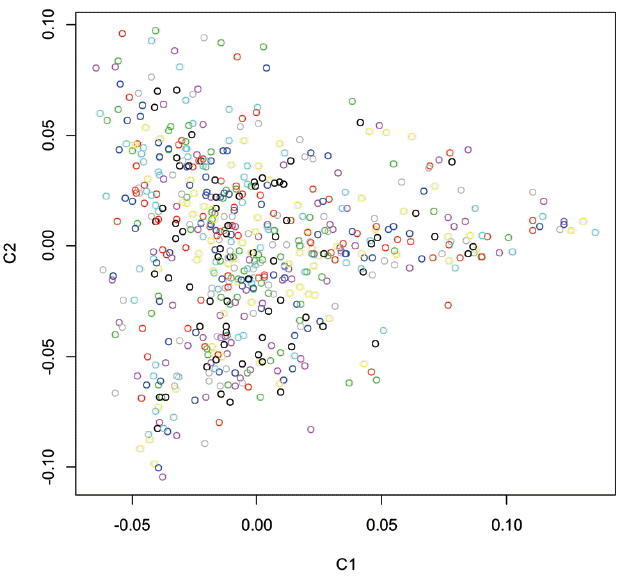
Principle component analysis (PCA) plot of population structure showing the top two principle components. C1: principle component 1; C2: principle component 2.
Most of the candidate genes we identified function in growth-related pathways, directly or indirectly. For example, SOGA1 participates in glucose production regulated by adiponectin, and was associated with the residual feed intake trait in another GWAS with Yorkshire boars [20,21]. Candidate gene ARHGAP24 is a key regulator of angiogenesis through its effects on endothelial cell migration, proliferation and capillary tube formation [22]. In our previous GWAS with Duroc male pigs, five suggestive SNPs, all associated with both D100 and ADG, were located within the ARHGAP24 gene [2]. This suggests that ARHGP24 is an important gene involved in growth. Furthermore, GO analysis indicates that most of the genes located close to the significant or suggestive SNPs are involved in lung development, respiratory system development, blood vessel morphogenesis, heart development and regulation of transcription (Table 4). Pathway analysis shows that candidate genes PLCB1 and MAP2K6 genes participate in the gonadotropin signaling pathway (GnRH) (Figure 3). A GWAS based on genetic, physiological and metabolomics data suggested that the gonadotropin signaling pathway is associated with divergent growth at the onset of puberty in cattle [23]. GnRH signaling also plays an important role in growth at the onset of puberty in mammals [24,25]. MAP2K6 plays an important role in adipocytes and glucose transporter expression [26–28] and PLCB1 is required for 3T3-L1 adipocyte and C2C12 myoblast differentiation [29,30]. These results suggest that the growth trait is affected in puberty through the GnRH signal pathway (MAP2K6 and PLCB1) in Yorkshire gilts. Other candidate genes are involved in the physiology of growth, such as GLI2 in myogenesis and osteoblast differentiation [31], NYAP2 in brain size, neurite elongation and neuronal morphogenesis [32], and ZFPM2 in adipogenesis [33].
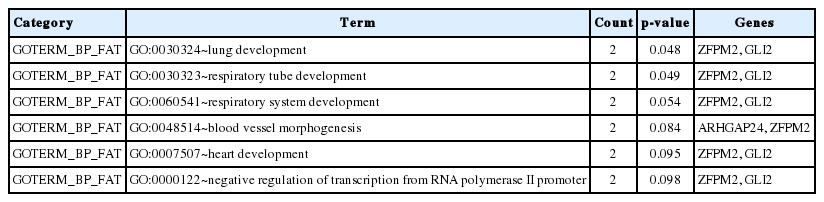
Gene ontology analysis of nearest genes
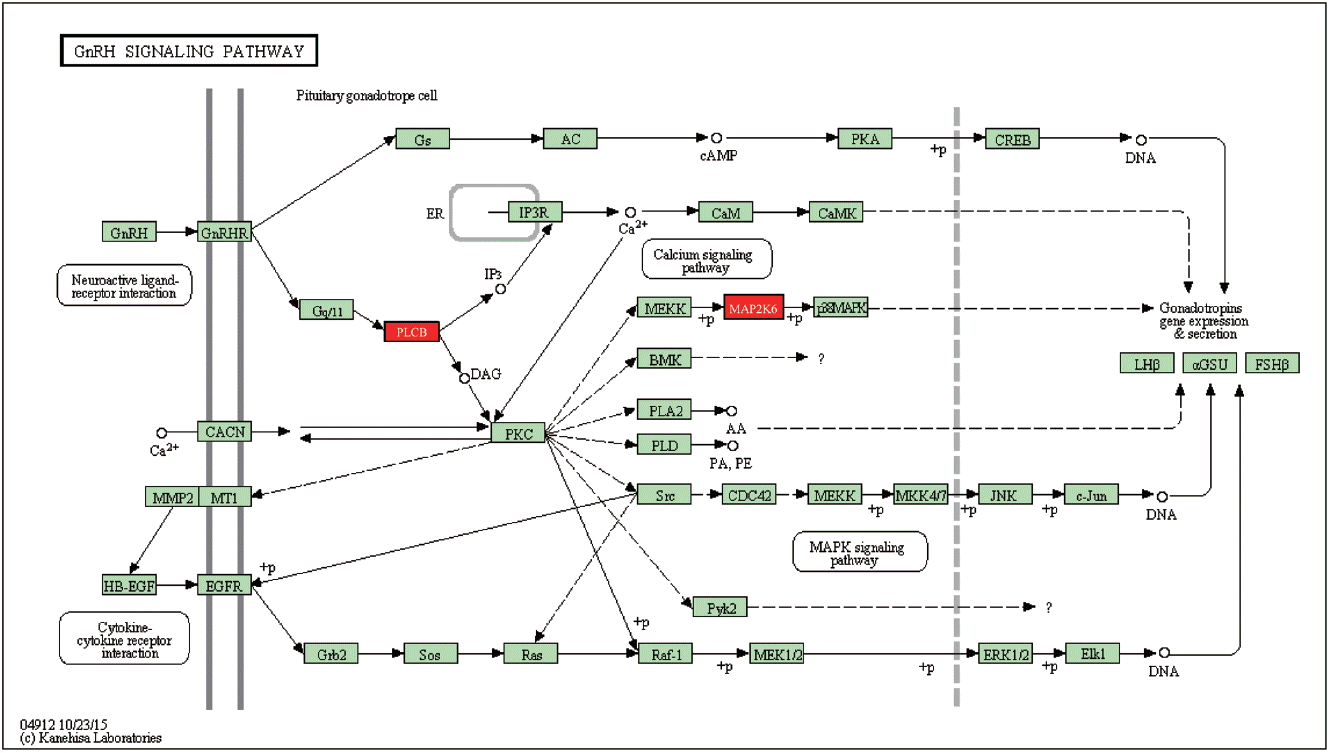
GnRH signaling pathway containing PLCB1 and MAP2K6. GnRH, gonadotropin-releasing hormone; PLCB1, phospholipase C beta 1; MAP2K6, mitogen activated protein kinase kinas.
Because the ADG and D100 or D115 traits measure similar (although not identical) growth stage periods, it is expected that some significant SNPs will associate with more than one trait [34], while each trait will be associated uniquely with specific SNPs. Our results agree, in part, with previous QTL mapping and GWA studies. For example, a QTL associated with ADG on SSC15 has been located in a region between 135.2 Mb and 149.8 Mb [35]. Our results identified two significant SNPs (INRA0050471 and MARC0061404) located at 140 Mb on the same chromosome. A region on SSC4 at approximately 34 Mb was associated in another GWAS with ADG in a Yorkshire population, in close proximity to the ZFPM2 gene [10]. Our study associated D115 with a significant SNP (ASGA001916) that is also close to this gene. Finally, our previous GWAS identified five suggestive SNPs, associated with both D100 and ADG, located within the ARHGAP24 gene in a Duroc male population [2]. In the present study, we identified a suggestive SNP (ALGA0113286) associated with D100, located close to the ARHGAP24 gene.
It is surprising that our results overlap only partially with our previous study. Possible reasons include: large numbers of causal variants with smaller effects are difficult to identify statistically [2], the pure pig population used in this study excluded many monomorphic SNPs, and incomplete power of different experimental designs [1].
In summary, using a panel of 54,148 SNPs, nine candidate genes were identified for the four growth-related traits D100 and D115 (days to 100 or 115 kg of body weight) and ADG100 and ADG115 (ADG between 30 and 100 or 115 kg). Further analysis revealed six significant and 12 suggestive SNPs, potentially causative mutations, in or near genes whose functions are plausibly growth-related. Further experimentation will be required to confirm the functions of these genes and elucidate the molecular mechanisms underlying growth traits.
ACKNOWLEDGMENTS
This work was supported by the Beijing San Yuan Breeding Technology CO., LTD Independent Project (SYZYZ20140007), Natural Science Foundation of China (NSFC, Grant 31372275) and Program for Changjiang Scholar and Innovation Research Team in University (IRT1191).
Notes
CONFLICT OF INTEREST
We certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.